Orthogonal Comparison of Analytical Methods by Theoretical Reconstruction from Bottom-up Assay Data
In the never-ending endeavor to produce stable and efficacious protein therapeutics, biopharmaceutical companies often employ numerous analytical techniques to characterize and quantify a drug candidate’s stability.
About This Article
Orthogonal Comparison of Analytical Methods by Theoretical Reconstruction from Bottom-up Assay Data
J Am Soc Mass Spectrom. 2021 Aug 4;32(8):2013-2018. doi: 10.1021/jasms.0c00433. Epub 2021 Mar 25.
Andrew C Nichols, Yong Joo Kil, Andrew Mahan, Bo Zhai, Robert Hepler, Kristen Nields, Hirsh Nanda, Eric Carlson, Marshall Bern
In the never-ending endeavor to produce stable and efficacious protein therapeutics, biopharmaceutical companies often employ numerous analytical techniques to characterize and quantify a drug candidate's stability. Mass spectrometry, due to the information-rich data it produces, is commonly used in its numerous configurations to ascertain chemical and structural stability.
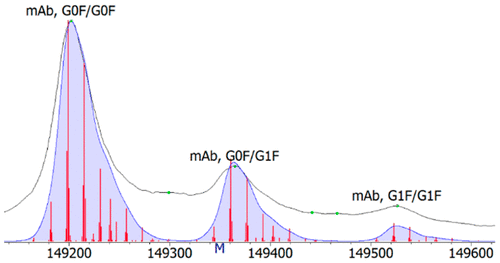
At issue is the comparison of the various configurations utilized, that is, comparing bottom-up methods such as proteolytic digest followed by reversed phase LC-MS with intact LC-MS methods. Similar issues also arise when using capillary isoelectric focusing to see how charge variants change over time, that is, monitoring the progression of charge altering modifications like deamidation. To this end, site-specific degradations as quantified from bottom-up methods like peptide mapping can be used to build reconstructions of both theoretical intact mass spectra as well as theoretical electropherograms. The result can then be superimposed over the experimental data to qualitatively, and perhaps quantitatively, evaluate differences.
In theory, if both experimental bottom-up data and intact data are accurate, the theoretical reconstruction produced from the bottom-up data should perfectly overlay with that of the experimental data. Valuable secondary information can also be ascertained from reconstructions, such as whether modifications are stochastic, as well as a detailed view of all possible combinations of modifications and their quantities used in the reconstruction. This comparison is also useful in determining unknown mass differences in deconvoluted intact protein spectra that may be a result of multiple modifications in combination. The comparison of data from alternate sources provides a holistic and more comprehensive view of the molecule under study.