Parsimonious Charge Deconvolution for Native Mass Spectrometry
A new “parsimonious” charge deconvolution algorithm well-suited to high-resolution native MS of intact glycoproteins and protein complexes.
About This Article
Parsimonious Charge Deconvolution for Native Mass Spectrometry
J Proteome Res. 2018 Mar 2;17(3):1216-1226. doi: 10.1021/acs.jproteome.7b00839. Epub 2018 Feb 8.
Marshall Bern, Tomislav Caval, Yong J Kil, Wilfred Tang, Christopher Becker, Eric Carlson, Doron Kletter, K Ilker Sen, Nicolas Galy, Dominique Hagemans, Vojtech Franc, Albert J R Heck
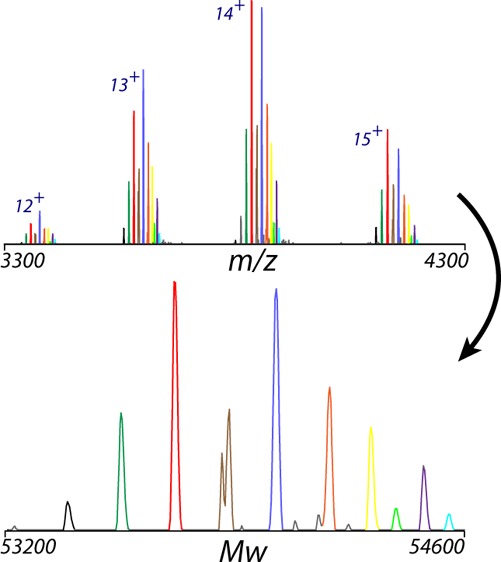
Charge deconvolution infers the mass from mass over charge (m/z) measurements in electrospray ionization mass spectra. When applied over a wide input m/z or broad target mass range, charge-deconvolution algorithms can produce artifacts, such as false masses at one-half or one-third of the correct mass. Indeed, a maximum entropy term in the objective function of MaxEnt, the most commonly used charge deconvolution algorithm, favors a deconvolved spectrum with many peaks over one with fewer peaks.
Here we describe a new "parsimonious" charge deconvolution algorithm that produces fewer artifacts. The algorithm is especially well-suited to high-resolution native mass spectrometry of intact glycoproteins and protein complexes. Deconvolution of native mass spectra poses special challenges due to salt and small molecule adducts, multimers, wide mass ranges, and fewer and lower charge states.
We demonstrate the performance of the new deconvolution algorithm on a range of samples. On the heavily glycosylated plasma properdin glycoprotein, the new algorithm could deconvolve monomer and dimer simultaneously and, when focused on the m/z range of the monomer, gave accurate and interpretable masses for glycoforms that had previously been analyzed manually using m/z peaks rather than deconvolved masses. On therapeutic antibodies, the new algorithm facilitated the analysis of extensions, truncations, and Fab glycosylation. The algorithm facilitates the use of native mass spectrometry for the qualitative and quantitative analysis of protein and protein assemblies.